As described in Chapter 8 of Bacterial Genetics and Genomics, a two-component regulatory system has a sensory protein in the membrane that autophorphorylates when triggered by its sensory signal and a regulatory protein that is phosphorylated to do its job adjusting gene expression. In this way, something sensed by the cell at its surface can influence the genes that are turned on or off and therefore respond to the stimulus.
Since this process is only required by the bacterial cell some of the time, we would not expect the components of a two-component regulatory system to be essential for the life of the cell. But, there are some studies that are showing essential regulatory protein components, suggesting another role for these proteins.
In Sinorhizobium meliloti, a soil bacterium particularly associated with legumes, the roles of the CenK histidine kinase sensor and the CenR response regulator were assessed. The CenKR two-component regulatory system has been shown to regulate srlA, a gene for a thioredoxin-like protein that is associated with high salt tolerance. The srlA gene is activated by the CenKR system, with CenR binding to an inverted repeat region in slrA’s promoter.
Photograph of the root nodules formed due to S. meliloti.
Interestingly, CenR is essential in S. meliloti, with the bacterial cells unable to survive the absence of this regulatory protein. However, its partner in the two-component regulatory system, CenK, is not essential. This suggested that CenR was either doing something important without being phosphorylated by CenK or was getting its phosphorylation elsewhere.
Using some clever single amino acid substitutions in the CenR protein, Freire et al (2024) were able to show that the essential function of this regulator did not require the protein to be phosphorylated.
Phosphorylation is required for regulation of SrlA, therefore this is not the gene that is involved in the essential function of CenR. Identifying this will require some more research into the regulatory networks in S. meliloti. This illustrates the importance of researching gene functions in the lab and not making too many assumptions about function based on the annotation of the sequences. Yes, there is a two-component regulatory system, but the regulatory protein part of this system may have another job in the cell.
It doesn’t take much to alter the sequence of a protein. Whilst some single nucleotide changes to the DNA sequence can be tolerated, others cause the gene to encode a different amino acid in the protein. Again, some of these changes to the amino acid sequence can be tolerated and generate no phenotypic outward change to the bacteria. But there are some changes that can have a big impact.
One such change has recently been reported in Burkholderia cepacia, Gram-negative opportunistic bacteria. In patients with compromised immune systems, such as people with cystic fibrosis and chronic granulomatous disease, B. cepacia can cause fatal pulmonary infections.
Some of these B. cepacia have a brown pigmentation from pyomelanin. This pyomelanin has been suggested to be involved in virulence and resistance of the bacteria to oxidative stress. However, not all infection-causing B. cepacia are pigmented, raising questions as to the role of the pigment in virulence.
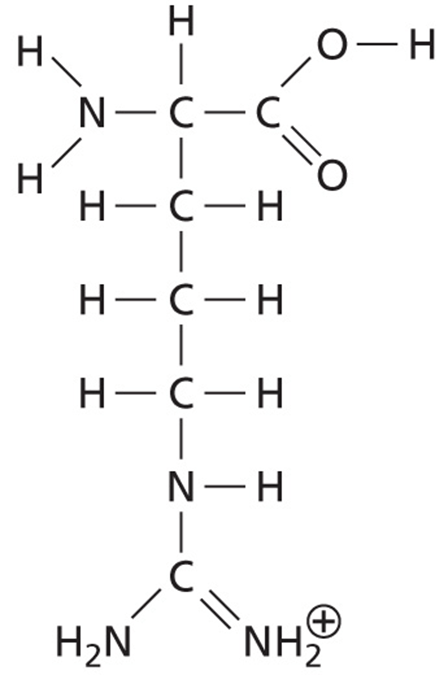
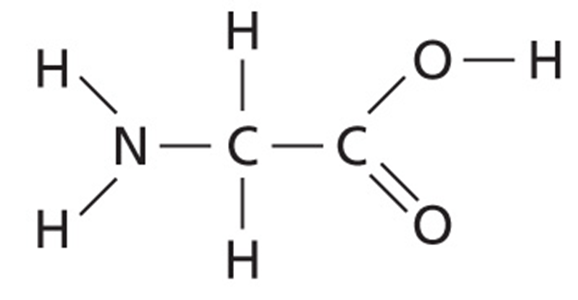
Interestingly, a change in a single amino acid in HmgA (homogentisate 1,2-dioxygenase) changes the pigmentation of the bacteria. In the pigmented bacteria, HmgA has an arginine at amino acid position 378. However, in the non-pigmented bacteria the amino acid at this position is glycine.
As we know from Bacterial Genetics and Genomics, Chapter 7, arginine is larger than glycine, having a positively charged side chain with four carbons, three nitrogens, and 11 hydrogens. Glycine, however, is the smallest and most flexible amino acid, with only a hydrogen for its side chain.
Figure of arginine (top) and glycine (bottom).
The two bacteria, one with arginine in HmgA and one with glycine in HmgA, were compared and found to have no difference in their resistance to oxidative stress by H2O2 and NO, despite the difference in pigmentation. There was also no difference in growth curves between the pigmented and non-pigmented bacteria. When investigated in a mouse model of infection, there was no difference in the virulence and infection outcome based on the HmgA sequence and the pigmentation of the bacteria. Therefore, there must be something other than the pyomelanin pigment at work. It could be that rather than pigment, or just pigment, there are other factors at work in different strains of B. cepacia that impact oxidative resistance and virulence.
Listeria monocytogenes and Escherichia coli are able to grow at low temperatures and from there be transmitted to humans and cause disease. Having antimicrobials that can work at low temperatures is important to breaking the transmission chain, particularly from food. However, most experiments on bacteria happen at human body temperature, 37°C. Thus, an antimicrobial that is effective at body temperature may not be as effective at refrigerator temperature.
Acidic electrolyzed water (AEW) is an antimicrobial that functions due to its low pH, high redox potential, and chlorine. It is considered to be an environmentally friendly way to reduce bacterial burden by disinfecting materials used in healthcare, in the food chain, and in other areas.
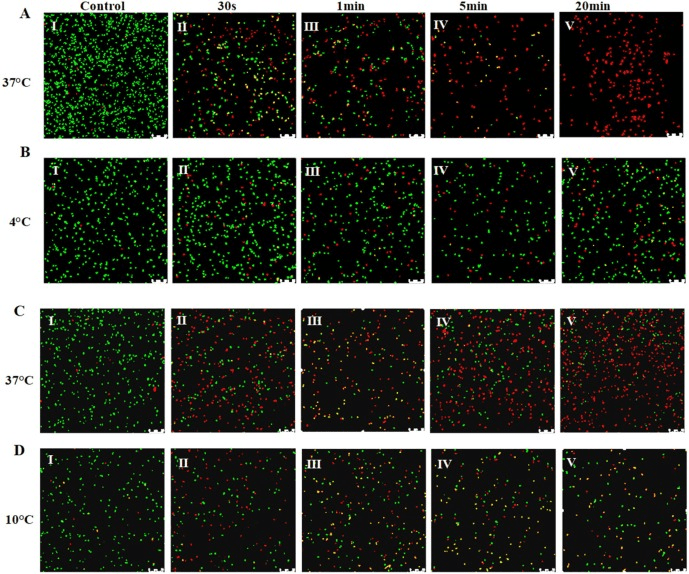
The utility of AEW at low temperature was not known and required some investigation. In this study, two bacterial species, L. monocytogenes and E. coli were grown at low temperature. L. monocytogenes was grown at 4°C and E. coli was grown at 10°C. These cultures were then treated with AEW. The effectiveness of disinfection was assessed for the bacterial cultures and the transcriptome was investigated using RNA-seq to see if there were differences in the gene regulation at low temperature that could be associated with AEW success.
Even with increased treatment time, AEW was found to be ineffective against cold grown bacterial cultures, suggesting that the bacterial regulation that enables growth at 4 – 10°C also provides resistance to the disinfection by AEW.
By observing the bacteria, it was revealed that the cells grown at 37°C had damaged cell membranes after AEW treatment and that this led to death of the cells. Bacteria grown at the lower temperatures had less membrane damage and were more likely to survive the treatment with AEW. Cell membrane damage was correlated with protein leakage from the cell.
Figure showing live bacteria in green and dead in red. Over time the 37C bacteria are killed by AEW and the 4C bacteria are not.
To simulate real-life situations, the effectiveness of AEW to disinfect salmon inoculated with L. monocytogenes or E. coli was assessed. This showed that the AEW was able to work on the salmon stored at 37°C but was less effective at lower temperatures required for long-term storage and transport of salmon to consumers. This could therefore lead to infection of humans via the stored salmon, even at 4°C.
The RNA-seq data from L. monocytogenes grown at 37°C, 4°C, and 4°C after AEW treatment was analyzed. This revealed a large number of differentially expressed genes. Cold-shock protein gene cspD is known from several bacterial studies to be involved in regulation in low temperatures. It is therefore not surprising to find this gene differentially regulated. It has led to adaptation of the L. monocytogenes to oxidative and osmotic stress caused by the AEW treatment. After AEW treatment the already upregulated cspD, due to growth at 4°C, was further upregulated, demonstrating that CspD is pivotal in resistance to the antimicrobial effects of acidic electrolyzed water.
These results show that our standard experimental conditions may not be revealing the complete picture. In our food cold chain there are instances where bacteria will be growing at the low storage temperatures of the food and therefore these bacteria need to be dealt with and experiments need to be done at these temperatures.
The regulation of gene expression means that genes are not ‘on’ all of the time, being transcribed and translated to make protein. The orchestration of gene expression is essential to the processes of the bacterial cell and can involve a number of mechanisms.
Often a regulatory protein is involved in the regulation of its own gene. This means that there is a feedback loop where transcription and translation of the gene circles back and regulates its own transcription and translation. Most often we see this happen directly on the promoter of the regulatory gene, where its own protein interacts with the promoter and either represses or activates its own expression. In fact, half of the regulators in E. coli are autoregulated.
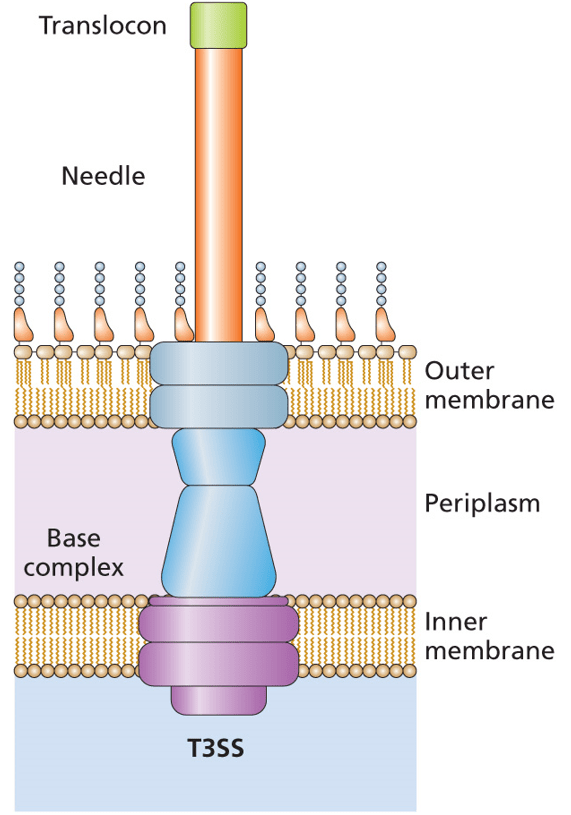
A recent publication describes things a bit differently. In Vibrio parahaemolyticus, the Type 3 Secretion System (T3SS) is regulated by the VtrB activator. These bacteria are resident in the marine environment, where they infect seafood that is part of our food chain. Eating raw or undercooked seafood can lead to an infection with V. parahaemolyticus. The Type 3 Secretion System (actually the second of two T3SSs) is the major virulence factor in these bacteria and it is activated by the VtrB regulatory protein. You can read more about Type 3 Secretion Systems in Chapter 9 and Chapter 13 of Bacterial Genetics and Genomics.
The expression of vtrB is activated by the bile acids that are secreted into the human intestinal tract. This causes the VtrA protein to bind to the promoter region of vtrB, activating its transcription and translation. It has been shown that this activation and expression of VtrB is crucial to infection in humans. Thus understanding it might give insights that could lead to better outcomes for V. parahaemolyticus infections.
The vtrB gene is autoregulated in an interesting way. Rather than VtrB interacting with the promoter of its own gene, the protein is involved in activation of the genes upstream (5’) of vtrB. There is an intrinsic transcriptional terminator between these upstream genes and vtrB. You can read more about intrinsic transcriptional terminators in Chapter 2 of Bacterial Genetics and Genomics.
VtrB causes the transcription of upstream genes to continue through to vtrB, despite the presence of the transcriptional terminator. This read-through transcription increases the expression of VtrB and thus increases the expression of the Type 3 Secretion System in the human body and the associated virulence.
This means that unlike the Type 3 Secretion Systems in species such as Salmonella, Shigella flexneri, and E. coli that autoregulate, with the regulatory protein working with its own promoter, the V. parahaemolyticus relies on read-through transcription. Assessment of the transcriptional terminator suggests that it is weak, with a low stability hairpin being formed, which is less likely to disengage the RNA polymerase. In addition, the transcriptional terminator is also very close (one base away) to the stop codon, which can also lead to read-through. A lesson to take from this is that autoregulation of a transcriptional regulator may be occurring, even if the regulator is not activating or repressing its own promoter. Check upstream of the regulatory protein gene. If the upstream genes are transcribed in the same direction and the transcriptional terminator between is weak, then maybe investigating read-through transcription will be worthwhile.
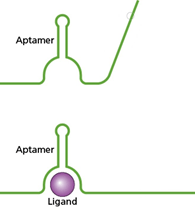
The specificity of antibody binding has been exploited for many years in a variety of technologies. Although perhaps less famous, aptamers also have high binding specificity for their targets and, being made of RNA or DNA rather than protein, are much smaller in size, can be modified more easily, can be more easily reproduced, and can be stored and delivered more easily than antibodies.
Figure 4.14 from Bacterial Genetics and Genomics. In the top panel, an Aptamer is depicted as a bent green line. In the bottom panel, the Aptamer has bound a target and changed conformation; the green line has changed in shape around a purple sphere representing a Ligand.
It is by virtue of these properties that aptamers have been explored for a range of biotechnology applications, including as tools for detection of bacterial contamination of foods for human consumption.
The food-borne pathogen Vibrio parahaemolyticus is the leading cause of seafood-associated bacterial gastroenteritis. All of the traditional methods of detection rely on bulky laboratory equipment and are time-consuming. In a paper by Jiang et al., 2021, the goal was to create an electrochemical aptasensor. This combined microfluidic technology with the specificity of binding in an aptamer to enable detection of the V. parahaemalyticus outside of a laboratory within 30 minutes. The paper demonstrates the sensitivity and specificity of their device and suggests that it could be adapted to detect other bacterial pathogens.
Photograph of raw seafood, a common source of transmission of V. parahaemolyticus to humans. Photograph from Photographer Sergy from Thailand.
Although we tend to think of Staphylococcus aureus perhaps in other contexts, it is also an important food-borne pathogen, particularly in the USA. To overcome time delays associated with traditional methods of detection of S. aureus using culturing, Yang and colleagues developed an aptamer-based technology that makes use of established portable detection platforms available in personal glucose meters. These hand-held devices are already readily available and reliable, so they made an ideal starting point for development of a portable bacterial detection device. The sensitivity of the aptamer biosensors were able to selectively detect S. aureus in food samples, demonstrating the usefulness of the portable technology.
Portable glucose meter icon image by By Laymik, UA. An icon drawing of a glucose meter, which has been used as part of an aptamer-based detection technology by Yang et al., 2021.
There is a study published in Analyst, a journal from the Royal Society of Chemistry, which investigated the contamination of powdered infant formula by the food-borne pathogen Cronobacter sakazakii. These bacteria cause meningitis, sepsis, and necrotizing enterocolitis in premature and immune-compromised infants. It is therefore vitally important that these bacteria do not end up in products for babies, like powdered infant formula. This particular study by Hye Ri Kim and colleagues, published in 2021, did not use the standard SELEX technique to develop its aptamers for detection of these pathogens. SELEX is ‘systematic evolution of ligands by exponential enrichment’ and has been used since 1990 to produce aptamers. Instead of SELEX, Kim et al. used a centrifugation-based partitioning method (CBPM), which produces target-specific aptamers in a shorter time-frame. Using this CBPM method, these researchers were able to isolate two aptamers against C. sakazakii and demonstrated that these could be used to efficiently detect the pathogen in powdered infant formula.
An icon drawing of a baby bottle beside a can of powdered infant formula with a scoop measure above it. Infant formula icon image by Chiara Rossi, IT
Sometimes the issue isn’t the bacteria, but rather the antibiotics we have been using to combat the bacteria. For example, an aptamer was developed by Komal Birader and colleagues that specifically detects the antibiotic oxytetracycline in milk. This antibiotic was used in veterinary practice, however it was banned due to potential side effects. This meant that it was necessary to develop a way to detect oxytetracycline in milk that was destined for human consumption. The detection method needed to be both affordable and one that could be used in the field. This team was able to modify the aptamer that they developed so that the presence of the antibiotic would result in a visual detection, making it ideally suitable for use in the field.
Much like antibodies have been used in a variety of ways that are well outside of their original purpose inside our bodies to fight off foreign invaders, aptamers are therefore being used in a range of ways that are beyond their original purpose. As explored in Chapter 4 of Bacterial Genetics and Genomics, aptamers can have a role in bacterial gene expression, but outside of the bacterial cell, scientists have found a whole range of uses for them, including detecting bacteria and antibiotics in our food.
One of the concepts that is discussed in Bacterial Genetics and Genomics is the difference between essential genes and accessory genes. The essential genes are those that are required for the bacteria to live and the accessory genes allow it to do something else that might be beneficial, but not essential. This is the difference between having all of the genes needed for cellular division and having the genes needed for a bacterial capsule. Being able to divide is an essential function and without it, the bacteria are not viable. The bacterial capsule may enable the bacteria to avoid the human immune system thus boosting its survival, but it is not essential. In this comparison, those that are essential and those that are accessory are determined within the bacteria itself, not by comparing between bacteria – even those of the same species.
This is an image from a microscope of bacteria expressing accessory genes for the bacterial capsule. Some bacterial cells are surrounded by a layer of extracellular polysaccharides that form the bacterial capsule. The capsule is visible here as the dark blue layer surrounding the bacterial cells. The capsule can help the bacteria avoid the immune system and resist drying. (This is from Bacterial Genetics and Genomics Figure 9.14, image courtesy of Dr Kari Lounatmaa/Science Photo Library.)
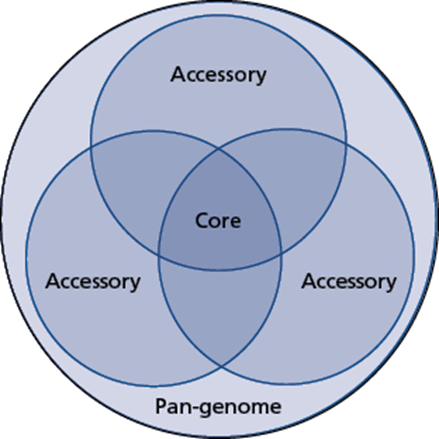
Another concept discussed is the difference between core genes and accessory genes. In this case, we are comparing the genes present between two different bacteria of the same species. The genes that are in common between the bacteria are core. Those that are not are accessory. This is often represented with a Venn diagram, showing all of the genes that are shared between two or more bacterial strains – the core genes – and those that are not and are accessory genes. Everything together is the pan-genome.
The core genome, accessory genome, and pan-genome. The core genome includes all of the genetic features that are included in all of the examples of a particular species. Each genome will, in addition, have other genetic features that contribute to the accessory genome. When all of the potential genes in a species are considered, everything from the core genome and all accessory genomes, even when not seen together in the same cell, this is the pan-genome. (This is from Bacterial Genetics and Genomics Figure 3.4.)
It is interesting that both of these concepts use the term accessory gene – something that could easily confuse students and researchers when discussing the nature of these genes. The point being that the genes in question are not essential to the function of the bacterial cells and quite often are not present in the genomes of all examples of that species.
In looking into this topic in the research literature, I came across a paper in PNAS (Proceedings of the National Academy of Sciences of the United States of America) by Bradley Poulsen and colleagues in Massachusetts, published in 2019. This article begins with a quite frank overview of what was believed to have been the promise of bacterial genome sequencing when it comes to the development of new antimicrobials. As they describe it, the revolution that was set to transform antibiotic discovery through the ability to identify essential genes and target them for new drugs, made possible from the first bacterial genome sequence in 1995, has just simply failed to materialize.
There are, of course, a number of problems that have arisen for which genomics is not entirely to blame. As noted in the PNAS paper, generally the bacterial membrane and efflux pump systems need to be overcome for any new drugs to be able to reach their targets. This is a strength of our bacterial foes, not a weakness of our genomic methods. Finding antimicrobials in some cases starts with screens of chemical libraries and improvements on these are often needed, which is again not a shortcoming of genomics. Lastly, there have been some successes in identification of new antibiotics, but often these are not broad-spectrum agents and therefore their development is abandoned or not picked up by pharmaceutical companies for further development and clinical trials due to the lack of profitability. This is again, not the fault of genomics.
However, the definition of essential genes does fall within genomics. In some cases, targets have been pursued that have not actually been essential, at least not in all species or in all strains of a species. This results in inhibitors having been developed to bacterial targets that are only effective against a sub-population of the pathogen, which is pretty much useless in a clinical setting.
In addition, essential genes can be essential for a particular set of circumstances. They can be essential depending on how the bacteria are being grown, for example, but if the bacteria are grown in different conditions those genes are no longer needed and would have otherwise been classified as accessory genes. These are ‘conditional essential genes’ – ones that are essential only under certain conditions. The bacteria may not encounter that condition or may be able to avoid being in that condition within the patient and therefore could avoid being cleared by the antibiotic developed to target the essential gene by not needing to use the gene and staying somewhere else in the body. It is quite common for genes to be essential in laboratory media, which is an artificial environment, but entirely different sets of genes to be needed for the various environments encountered in the body, like the gut, the blood, the mucosal surfaces, and inside cells.
Poulsen et al. proposed in their 2019 PNAS paper that antibiotic drug discovery could be improved through focusing on core essential genes – those that are shared amongst all of the sequenced genomes of a species AND that could be demonstrated to be essential in a range of different conditions that mimic the human body, not just one. In their proof-of-concept investigation, they chose to look at Pseudomonas aeruginosa due to the urgent need for novel antibiotics.
To find essential genes, they used transposon insertion sequencing. This is known as Tn-Seq, TIS, INseq, HITS, and TraDIS. Two P. aeruginosa laboratory strains were mutated in this way, strain PA14 and PAO1. Eight other strains were also investigated under five growth conditions and a statistical method called Finding Tn-Seq Essential genes (FiTnEss) was used to identify the core essential genes from the data collected.
In the end, the conclusion of the authors is that it is not genomics that has failed us in providing answers that we need for developing new antibiotics to combat the growing threat of antimicrobial resistant infections. It is rather our own inability to use genomics to its best advantage and to design experiments to best identify the genes that need to be targeted by novel antimicrobials. Through the advances in genomic technologies, it is now possible to rapidly and inexpensively sequence and re-sequence bacterial genomes, which was not possible in the 1990’s, 2000’s, and even 2010’s. At the start of the 2020’s, we are able to use the advances of genomics to do the types of experiments we have wanted to do, but have often been out of reach due to the expense and scope that would have been out of reach previously.
Therefore, the quest for core essential genes is has only just begun.
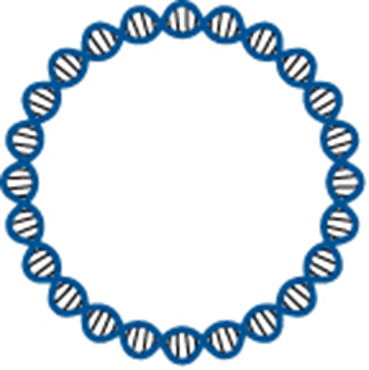
When we first started sequencing bacterial genomes, back in the 1990s, the goal was to ‘finish’, to generate a complete, closed, circularized chromosomal sequence that as accurately as possible reflected what would be seen in the bacterial cell. This process often took months, if not years, and required extensive laboratory work. It generated sequences such as the first genome sequence of a free-living organism, the sequences of model organisms E. coli and Bacillus subtilis, and the first comparative genome sequence analysis.
Graphic design of the DNA double helix as a circular chromosome. Not to true proportions. Cropped from Figure 2.3 of Bacterial Genetics and Genomics, Chapter 2: Genes.
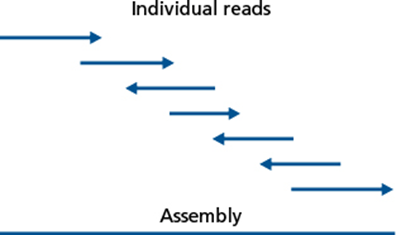
As time went on and sequencing technology changed, it became faster and less expensive to do whole genome sequencing, yet the challenge of finishing the genome sequence into a closed chromosome remained. It became standard to analyze incomplete sequence data as contigs, contiguous strings of sequence data that had been assembled based on localized homology from raw sequence reads.
Figure illustrating the assembly of individual sequencing reads generated by sequencing technologies into an assembled contig, ready for analysis. Figure 17.2 of Bacterial Genetics and Genomics, Chapter 17: Genome Analysis Techniques.
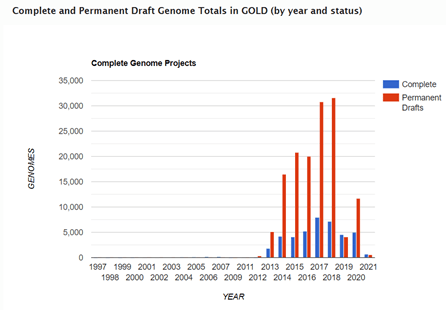
Although these contigs ranged in lengths and the number of contigs per sequenced genome also varied, due to features within the chromosome such as repetitive and within genome homologous regions and repeats, assemblies did not generate closed sequences. Therefore, as time went on in the 2000’s and 2010’s, the number of genome sequences in the public databases such as GOLD (Genomes Online Database) referred to as ‘permanent drafts’ continue to grow exponentially.
Graph generated by the Genomes Online Database from available sequence data in GOLD. Complete are finished to finalized chromosomes, while permanent drafts will remain in contigs because no further sequencing is planned for that sample to improve the sequence data or close the gaps.
Since this data in the ‘permanent drafts’ is not assembled into the final chromosome and is potentially incomplete or contains sequencing errors, someone new to the field or unfamiliar with genomics may initially assume that the draft nature of the data means that it is still in progress and will eventually be completed. For the vast majority of projects, this is not the case. This is simply because, genome sequencing projects have learned to adapt to the data that is being generated and are able to use this draft data, assembled into contigs, to answer the scientific questions that are being posed.
There are many examples of this in the literature dating back just shy of 20 years, to the earliest publications using next-generation sequencing technologies. In this blog, I am going to highlight a paper by Joon Liang Tan et al. from 2020, which has investigated 15 newly generated genome sequences of Mycobacterium tuberculosis from Malaysia, using MiSeq by Illumina. This paper is a good example of the type of analysis that can be done with genome sequences that are in contigs.
Scientific Data journal article by Joon Liang Tan, et al., 2020, ‘Genome sequence analysis of multidrug-resistant Mycobacterium tuberculosis from Malaysia’. Screenshot of the title with author list and Abstract.
As explained in the paper, multidrug resistant M. tuberculosis (MDRTB) is still relatively rare in Malaysia (1.5% of cases) and M. tuberculosis (TB) itself is present at an incidence of 92 per 100,000 population as of 2019. In this study, 15 MDRTB archive isolates from patients from 2009 to 2012 from the University of Malaya Medical Center were whole genome sequenced using MiSeq.
The raw sequence data underwent quality control, with poor quality reads being removed before assembly. The quality reads were then assembled into contigs, which were polished to improve the accuracy of the sequence data. This draft genome sequence data, in contigs, was then annotated using Prokka. The Prokka annotation data was used to identify core and accessory genomes with Roary. The Prokka and Roary data together fed into the Piggy analysis for intergenic region prediction. Lineages were predicted using the TB Profiler Web server. Based on the Prokka annotation of predicted protein encoding genes, proteins sequences were submitted to the Comprehensive Antibiotic Resistance Database. The 15 MDRTB draft genome sequences were also compared against the collection of MDRTB genome sequences at the Broad Institute.
Often, such projects compare draft genome sequence data to an established reference genome. This relies upon there being a complete, closed, circularized genome sequence that is representative for the species. This means that at some point in the past someone had to do the hard work of finishing the genome sequence. In this case, the reference genome sequence used was M. tuberculosis H37Rv. Each of the 15 MDRTB draft genome sequences was compared against this reference genome. This draft sequence data is in fragments, the assembled into contigs, ranging from 149 contigs to 439 contigs. It is not unusual for draft genome sequences to have over 100 contigs. It is quite good for a draft bacterial sequence to have under 100 contigs or better yet under 50. The contigs are aligned against the reference genome sequence to determine how much similarity there is between the newly sequenced bacterial genome and the reference bacterial genome. In this case, an average of 99.26% of the M. tuberculosis H37Rv reference genome sequence was covered by the 15 Malaysian MDRTB sequences. This means that almost everything found in H37Rv could be found in these Malaysian isolates. Focusing just on the core gene families that were identified in the 15 MDRTB isolate sequences by Roary, about 97% were also present in the reference genome sequence TB H37Rv. This means that most of the identified genes that are common between the Malaysia isolates are also found in the reference genome, but a few are not. Of those that are not, most are hypothetical (predicted by the software, but where function is not known).
Shifting to analysis of the intergenic regions, there were 2,172 to 2,288 regions between the CDSs in the 15 MDRTB isolate sequences. Of these, 1,365 were found in all of the 15 MDRTB isolate sequences and 1,453 were present in at least two of the isolates. Of the intergenic regions, 974 were found in only one of the 15 MDRTB isolate sequences. These sequences between the CDSs were one of the main contributing factors to differences between the isolates and between the isolates and the reference strain M. tuberculosis strain H37Rv.
Seven of the 15 MDRTB isolate sequences were classified as part of the East Asian Lineage 2.2.1 and four part of East Asian Lineage 2.1. Two isolates were determined to be part of the Indo-Oceanic Lineage 1.1.3 and one included in the Euro-American Lineage 4.3.4.1. The final isolate was of Lineage 1.2.2. The SNP differences between each isolate indicated that they were epidemiologically distinct and not related to one another. I think it would have been nice to see a phylogenetic tree with the data from these isolates, to put the Malaysian TB into visual context with sequenced TB from the rest of the world, but this publication does not include any figures.
The investigation also conducted a detailed analysis of the antimicrobial resistance determinants in the genetic data. Importantly, these analyses showed that there is no evidence of XDRTB (extensively drug-resistant tuberculosis) because there wasn’t resistance to: 1. first-line anti-TB drugs (isoniazid; pyrazinamide; streptomycin; rifampin; and ethambutol) and; 2. a fluoroquinolone and; 3. at least one second-line drug (amikacin, kanamycin, or capreomycin). To be classed as XDRTB, all three criteria need to be met. The isolates are not only phenotypically not XDRTB (they did not display resistance in lab tests), but they also are not genotypically XDRTB (they don’t have the genetic features needed to satisfy all three criteria). Due to regulation and mutation, it is possible for bacteria to carry a gene, but not express its phenotype. However, if evidence of the genetic markers for XDRTB were identified, this could be concerning. Fortunately, there was none.
The authors highlight the importance not only of monitoring antibiograms of isolates for epidemiology, but also regular sequencing to evaluate the distribution of lineages and genetic basis for observed resistance.
This information was all gained, vitally, from genome sequence data that was not ‘finished’. Indeed, one of the genomes was in quite a lot of contigs (439!), yet meaningful information was extracted from these pieces of the whole chromosomal puzzle. Whilst a complete chromosome can be ideal for answering a range of research questions, there is a great deal that can be done with the vast quantities of incomplete, draft data that is available in the public databases and is continuing to be generated.
For these blogs I have not been including the wording of the end of chapter questions from Bacterial Genetics and Genomics. Instead, I have blogged about the general theme of these question, often highlighting a research article on the topic.
However, today (8th March 2021) is International Women’s Day and the very last self-study end of chapter question in the book is very relevant:
“Esther Lederberg discovered lambda (λ) bacteriophages and described lysogeny. She also made other contributions to microbiology and microbial techniques that contributed to bacterial genetics and genomics. The study of λ provided insight that was extrapolated across the field of genetics. Explore and discuss the contributions made by Esther Lederberg and at least one other scientist who made important contributions, but may not be well known, perhaps due to gender or race.”
Prof. Lederberg is mentioned more than once in the book, having been instrumental in the discovery of not only lambda (λ) bacteriophages, but also description of F factor in bacteria and development of the replica plating technique. I encourage you to look for more information about Prof. Lederberg and her various contributions to microbiology.
Another woman I would like to discuss in this blog is Jane Hinton. There are some bacterial growth media with interesting names, including Mueller-Hinton agar as well as a range of other broths and agars that are all obviously named after someone. We don’t often think about the people who took their time and effort to develop these valuable and essential resources that enable us to do the fundamental aspect of our work – culturing bacteria. Most of my research is on Neisseria gonorrhoeae and Jane Hinton was involved in creating the Mueller-Hinton media that was instrumental in making culturing of N. gonorrhoeae practical.
Portrait photograph of Dr. Jane Hinton from The 1949 Scalpel, yearbook of the Senior Class, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, Pennsylvania
Dr. Hinton was the daughter of Prof. William Augustus Hinton, the first African-American professor at Harvard University and the first African-American author of a textbook. In 1931, he developed a Medical Laboratory Techniques course that was open to women, which led to Jane Hinton working with John Howard Mueller at Harvard on the creation of a media for culturing N. gonorrhoeae and Neisseria meningitidis. The Mueller-Hinton agar became the standard for antibiotic susceptibility testing, due to their incorporation of starch in the media, which enhanced growth and produced reliable antimicrobial testing results, and the transparent nature of the media, making the plates easier to read than opaque chocolate agar media, as well as easier to make.
In 1949, Dr. Jane Hinton graduated from the University of Pennsylvania as a Doctor of Veterinary Medicine. She and Alfreda Johnson Webb, graduating from the Tuskegee Institute, were the first two African-American women veterinarians.
When you are next in the lab or writing up a method and come across a name for growth media or a technique, perhaps take a minute to look into the history behind the name and the microbiology discoveries that went into what we think of as commonplace today. And remember that our discoveries and inventions made by women working in microbiology today may be historic events to someone in the future.
It has long been a defining difference that bacterial cells, like E. coli, have coupled transcription-translation and eukaryotic cells, like animals and plants, make their mRNA in the nucleus and their proteins in the cytoplasm. This is mostly the case. Mammals did break the rules and got found out a few years ago (see Coupled Transcription and Translation within Nuclei of Mammalian Cells). But, it seemed clear that the proteins made in the mammalian nucleus weren’t the main process for these cells and that bacterial cells were the clear leaders in doing coupled transcription-translation.
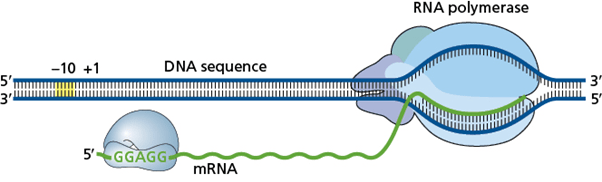
Bacterial coupled transcription-translation. RNA polymerase generates mRNA in transcription from the DNA sequence template. This mRNA is translated by ribosomes, which follow closely behind the RNA polymerase.
The transcription-translation complex (TTC) is the physical association of the RNA polymerase (RNAP), which is transcribing the DNA into mRNA, and the ribosome (containing rRNA and ribosomal proteins), which is translating mRNA into protein with the help of tRNAs carrying amino acids. The TTC is also known as the expressome.
It’s been discovered that bacteria have been breaking the rules as well. As we often find, when we try to define our world, we will come across exceptions to our rules and classifications, and we need to be able to reappraise our understanding as we learn more and do more research. This is one such example. Coupled transcription-translation is an ideal example from microbiology. A lot of what we know in microbiology, especially insights gained in early microbiology, is based on research in E. coli that was extrapolated across to other bacterial species. The more we learn about other bacterial species, the more we learn about how diverse they are and how much they do not do things the same as E. coli, like coupled transcription-translation.
In August of 2020, Grace E. Johnson, Jean-Benoît Lalanne, Michelle L. Peters, and Gene-Wei Li published research focused on the Gram-positive model species Bacillus subtilis that challenged the coupled transcription-translation paradigm that had been established 50 years earlier in E. coli, a model Gram-negative bacterial species.
Johnson et al., Nature 2020 Functionally uncoupled transcription-translation in Bacillus subtilis.
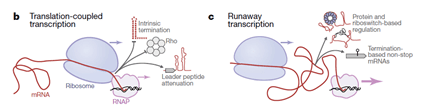
The B. subtilis RNAP was shown to transcribe DNA into mRNA much faster than the ribosomes can progress along the mRNA to conduct translation. This ‘runaway transcription’ means that the RNAP and ribosome are not coupled in transcription-translation, with RNAP moving at nearly twice the speed and leaving the translation machinery in its wake.
The differences between E. coli and B. subtilis with regards to the association between RNAP and the first ribosome conducting translation mean that there are also other differences. B. subtilis tends to rely less on Rho-dependent transcriptional termination and to have a greater prevalence of riboswitches in the mRNA leader sequences. Looking at these features, the researchers were able to suggest that ‘runaway transcription’ is not limited to B. subtilis and that there should now be two models of bacterial transcription and translation: translation-coupled transcription and runaway transcription.
From Johnson et al., Nature 2020, Figure 4b&c. Two models for bacterial transcription and translation, depending on species. On the left (b) is translation-coupled transcription, also known as coupled transcription-translation, in which RNAP is followed closely by ribosomes, as described first in E. coli. Characteristic here is intrinsic termination, Rho-dependent termination, and leader peptide attenuation. On the right (c) is runaway transcription in which RNAP runs much faster than the ribosome that follows, as described first in B. subtilis. Characteristic here is protein and riboswitch-based regulation, and termination-based non-stop mRNAs.
There is some further reading about insights into transcription – translation in bacteria, including coupled, uncoupled, and collided E. coli expressome states in a Nature Microbiology Reviews Research Highlight article.
The Johnson et al., 2020 contribution to our wider understanding of transcription and translation in bacteria establishes a second model of ‘runaway transcription’, which is a common feature of some bacterial species, rather than coupled transcription-translation. This is an important insight into bacterial genetics and genomics and a reminder of the diversity of bacterial species.
I recently attended and was an invited speaker, on-line, at a conference that had a topic about short-and long-read sequencing technologies. The various talks and the panel discussion that I participated in looked at the advantages of both short-read sequencing and long-read sequencing for what they can bring to research and to the clinical setting. The conference was Oxford Global NextGen Omics UK, which runs yearly, usually in London.
Following the conference, I decided that I wanted to blog about a study that had used sequencing to do something interesting. I found this paper, by Ovokeraye Achinike-Oduaran and colleagues at the University of the Witwatersrand, which describes a microbiome study from South Africa. Although the paper does not do whole genome sequencing of the microbiome (it is using 16S rRNA microbiome sequencing), it does overturn longstanding assumptions to develop its experimental design to investigate the gut microbiome.
Graphic in pink of the human small and large intestine overlaid with graphics of larger-than-life microbes. CC
Firstly, there is an issue in a lot of our human genomic data, microbial genome data, and metagenomic data. The vast majority of it has come from Western samples and from white patients. Although this may be changing, as with many things, change is slow. We cannot extrapolate information from the data we have to the whole, if we are lacking information from segments of that whole. In bacteriology, we have relied for a long time on extrapolating the knowledge gained by studying E. coli to other bacterial species, only to discover that many of these other species do things differently. We need more data about other bacterial species and about our own.
Secondly, it is important to remember that Africa is vast and varied. It is important to stop thinking of Africa as one homogeneous population and to remember that the people, the cultures, and the ecosystems and environments vary considerably across the continent. People living in different States in the USA have very different cultures, different weather conditions, and often different accents. In addition to that, they share the same continent with people in Canada and Mexico, who also have their own characteristic States and Provinces. The North American continent is diverse and so too is the continent of Africa.
Thirdly, may studies that have previously looked at the microbiome of humans in Africa have focused on those from populations that live in what Western cultures would consider to be “extreme” conditions, living in rural hunter-gatherer societies or agricultural populations. These studies have been vital in understanding traditional African population microbiomes and, in particular, the microbiomes of the populations that were studied. However, there has been a shift in the last 50 years or so to a more industrialized and sedentary lifestyle in sub-Saharan Africa. Therefore, these transition populations warrant investigation as well. There are a growing number of supermarkets and fast food outlets with trends toward Westernized processed and animal-based food products. As seen in other cultures, there are also associated reports of increases in obesity and decreases in physical activity across 24 African countries.
In this investigation, two South African populations are investigated, one urban and one transitioning rural. No assumptions are made about the ability to extrapolate findings from non-South African populations onto this community. The gut microbiomes here may be very different to elsewhere in the world, therefore necessitating investigation. Here there is an ongoing transition epidemiologically, which makes it a fascinating community to study, as the gut microbiome may be in transition as the dietary lifestyles of the people change.
The microbiota landscape of obese and lean female individuals from South Africa, chosen from diverse ethnolinguistic groups, were chosen for this investigation. It is noteworthy that even within the study, it is recognized that there is a wealth of diversity within South Africa, both in terms of the dietary transitions and also in terms of ethnolinguistic groups. This is a pilot study that is part of the Human Heredity and Health in Africa (H3Africa) initiative.
Although both cohorts in this study are from South Africa, they are 300 miles (483 km) apart and represent different lifestyles and diets. One group of females, ranging in age from 43 to 72 years, was from Bushbuckridge, a rural community. Here the researchers worked extensively with a community advisory group to ensure their research was performed in a sensitive and respectful manner that was clear and engaging with the community it was seeking to understand.
Photograph of Bushbuckridge in South Africa, a rural community. CC
The other group of females, ranging from age 43 to 64 years, was from the urban Soweto area, where the median BMI was 36.52 compared to 31.17 in the Bushbuckridge cohort.
Photograph of Soweto, South Africa, an urban community. CC
Benefitting from the data of previous microbiome studies, this investigation was able to reveal that their cohorts’ gut microbiomes had features of both taxa that tended to be found in Western microbiomes and in non-Western microbiomes, which has captured the transition state of the human gut during this transition period in the diets of this population. Some of the genera typical of hunter-gatherer societies are present, but so too are genera typical of people who consume a Western diet.
Given the issues of obesity in many Western societies and the growing issues of obesity in parts of Africa, particularly were Western culinary cultures are being adopted, Oduaran et al., (2020) specifically compared the obese and lean females within their study. They determined that there were no statistically significant within site differences for the Soweto cohort. Some differentially abundant taxa were observed in obese samples from Bushbuckridge, including association of Oscillibacter, which has been reported to be associated with obesity in a European cohort.
Photograph of Ovokeraye Achinike-Oduaran, lead author of the research study being discussed in this blog.
This study is an important pilot investigation. It shows that these intermediate states of human gut microbiomes are present in the current populations in South Africa, both in the urban setting and rural communities. The consequences of this mixed gut microbiome on human health is unknown, however through preliminary sequencing investigations such as this, the groundwork is set to be able to do more with larger cohort groups in cooperation with communities that are under-represented in our current research datasets. Future research will also be able to capture more sequence data information using enhanced sequencing technologies, perhaps in the not too distant future using robust long read sequencing on its own to generate high quality metagenomic data routinely.
This and other studies being conducted by researchers that are looking at under-represented populations of humans and under-studied bacterial species are important. We must not lose sight of the wonderous diversity of our planet that provides us with ever more to learn as biologists.













{kind=link}
{kind=link}