Bacterial Genetics and Genomics book Discussion Topic: Chapter 17, question 14
When we first started sequencing bacterial genomes, back in the 1990s, the goal was to ‘finish’, to generate a complete, closed, circularized chromosomal sequence that as accurately as possible reflected what would be seen in the bacterial cell. This process often took months, if not years, and required extensive laboratory work. It generated sequences such as the first genome sequence of a free-living organism, the sequences of model organisms E. coli and Bacillus subtilis, and the first comparative genome sequence analysis.
As time went on and sequencing technology changed, it became faster and less expensive to do whole genome sequencing, yet the challenge of finishing the genome sequence into a closed chromosome remained. It became standard to analyze incomplete sequence data as contigs, contiguous strings of sequence data that had been assembled based on localized homology from raw sequence reads.
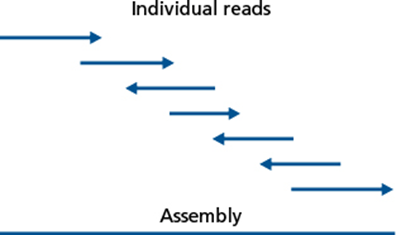
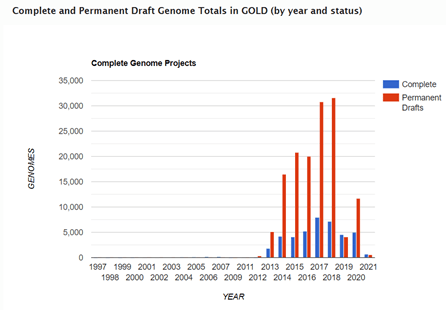
Although these contigs ranged in lengths and the number of contigs per sequenced genome also varied, due to features within the chromosome such as repetitive and within genome homologous regions and repeats, assemblies did not generate closed sequences. Therefore, as time went on in the 2000’s and 2010’s, the number of genome sequences in the public databases such as GOLD (Genomes Online Database) referred to as ‘permanent drafts’ continue to grow exponentially.
Since this data in the ‘permanent drafts’ is not assembled into the final chromosome and is potentially incomplete or contains sequencing errors, someone new to the field or unfamiliar with genomics may initially assume that the draft nature of the data means that it is still in progress and will eventually be completed. For the vast majority of projects, this is not the case. This is simply because, genome sequencing projects have learned to adapt to the data that is being generated and are able to use this draft data, assembled into contigs, to answer the scientific questions that are being posed.
There are many examples of this in the literature dating back just shy of 20 years, to the earliest publications using next-generation sequencing technologies. In this blog, I am going to highlight a paper by Joon Liang Tan et al. from 2020, which has investigated 15 newly generated genome sequences of Mycobacterium tuberculosis from Malaysia, using MiSeq by Illumina. This paper is a good example of the type of analysis that can be done with genome sequences that are in contigs.

As explained in the paper, multidrug resistant M. tuberculosis (MDRTB) is still relatively rare in Malaysia (1.5% of cases) and M. tuberculosis (TB) itself is present at an incidence of 92 per 100,000 population as of 2019. In this study, 15 MDRTB archive isolates from patients from 2009 to 2012 from the University of Malaya Medical Center were whole genome sequenced using MiSeq.
The raw sequence data underwent quality control, with poor quality reads being removed before assembly. The quality reads were then assembled into contigs, which were polished to improve the accuracy of the sequence data. This draft genome sequence data, in contigs, was then annotated using Prokka. The Prokka annotation data was used to identify core and accessory genomes with Roary. The Prokka and Roary data together fed into the Piggy analysis for intergenic region prediction. Lineages were predicted using the TB Profiler Web server. Based on the Prokka annotation of predicted protein encoding genes, proteins sequences were submitted to the Comprehensive Antibiotic Resistance Database. The 15 MDRTB draft genome sequences were also compared against the collection of MDRTB genome sequences at the Broad Institute.
Often, such projects compare draft genome sequence data to an established reference genome. This relies upon there being a complete, closed, circularized genome sequence that is representative for the species. This means that at some point in the past someone had to do the hard work of finishing the genome sequence. In this case, the reference genome sequence used was M. tuberculosis H37Rv. Each of the 15 MDRTB draft genome sequences was compared against this reference genome. This draft sequence data is in fragments, the assembled into contigs, ranging from 149 contigs to 439 contigs. It is not unusual for draft genome sequences to have over 100 contigs. It is quite good for a draft bacterial sequence to have under 100 contigs or better yet under 50. The contigs are aligned against the reference genome sequence to determine how much similarity there is between the newly sequenced bacterial genome and the reference bacterial genome. In this case, an average of 99.26% of the M. tuberculosis H37Rv reference genome sequence was covered by the 15 Malaysian MDRTB sequences. This means that almost everything found in H37Rv could be found in these Malaysian isolates. Focusing just on the core gene families that were identified in the 15 MDRTB isolate sequences by Roary, about 97% were also present in the reference genome sequence TB H37Rv. This means that most of the identified genes that are common between the Malaysia isolates are also found in the reference genome, but a few are not. Of those that are not, most are hypothetical (predicted by the software, but where function is not known).
Shifting to analysis of the intergenic regions, there were 2,172 to 2,288 regions between the CDSs in the 15 MDRTB isolate sequences. Of these, 1,365 were found in all of the 15 MDRTB isolate sequences and 1,453 were present in at least two of the isolates. Of the intergenic regions, 974 were found in only one of the 15 MDRTB isolate sequences. These sequences between the CDSs were one of the main contributing factors to differences between the isolates and between the isolates and the reference strain M. tuberculosis strain H37Rv.
Seven of the 15 MDRTB isolate sequences were classified as part of the East Asian Lineage 2.2.1 and four part of East Asian Lineage 2.1. Two isolates were determined to be part of the Indo-Oceanic Lineage 1.1.3 and one included in the Euro-American Lineage 4.3.4.1. The final isolate was of Lineage 1.2.2. The SNP differences between each isolate indicated that they were epidemiologically distinct and not related to one another. I think it would have been nice to see a phylogenetic tree with the data from these isolates, to put the Malaysian TB into visual context with sequenced TB from the rest of the world, but this publication does not include any figures.
The investigation also conducted a detailed analysis of the antimicrobial resistance determinants in the genetic data. Importantly, these analyses showed that there is no evidence of XDRTB (extensively drug-resistant tuberculosis) because there wasn’t resistance to: 1. first-line anti-TB drugs (isoniazid; pyrazinamide; streptomycin; rifampin; and ethambutol) and; 2. a fluoroquinolone and; 3. at least one second-line drug (amikacin, kanamycin, or capreomycin). To be classed as XDRTB, all three criteria need to be met. The isolates are not only phenotypically not XDRTB (they did not display resistance in lab tests), but they also are not genotypically XDRTB (they don’t have the genetic features needed to satisfy all three criteria). Due to regulation and mutation, it is possible for bacteria to carry a gene, but not express its phenotype. However, if evidence of the genetic markers for XDRTB were identified, this could be concerning. Fortunately, there was none.
The authors highlight the importance not only of monitoring antibiograms of isolates for epidemiology, but also regular sequencing to evaluate the distribution of lineages and genetic basis for observed resistance.
This information was all gained, vitally, from genome sequence data that was not ‘finished’. Indeed, one of the genomes was in quite a lot of contigs (439!), yet meaningful information was extracted from these pieces of the whole chromosomal puzzle. Whilst a complete chromosome can be ideal for answering a range of research questions, there is a great deal that can be done with the vast quantities of incomplete, draft data that is available in the public databases and is continuing to be generated.
