Bacterial Genetics and Genomics book Discussion Topic: Chapter 16, question 15
For this blog, I have decided to look at the Discussion Topic from Bacterial Genetics and Genomics, Chapter 16, question 15, which discusses restriction enzymes and encourages us to try finding digest sites for these enzymes ourselves in a gene of interest, using on-line tools. In the spirit of the last few blogs (see Doing research and making discoveries outside of the lab and More research outside of the lab, protein structure predictions), since many of us are working at home during the pandemic, either exclusively or with limited access to our labs, I have decided to take an approach that uses a minimum of tools to achieve the final goal. This means that regardless of your set up on your computer, you should be able to complete the task of identifying two restriction enzyme recognition sites, the places where the enzyme would digest the DNA. We want enzymes that would generate sticky ends and that will cut only once in the gene sequence, with one enzyme cutting at the beginning and one at the end of our gene of interest.
I’m going to use the same hypothetical gene that I investigated in the previous blogs. So far, I have been using the predicted protein sequence. This is easily extracted out of an annotation. Annotation files are plain text files and can be opened with a text editor file. I recommend Notepad for those using Windows because Notepad doesn’t add any formatting; a word processing programme like Word does add formatting, even to plain text files, so keep it simple with something like Notepad that opens .txt type files when you want to open FASTA and annotation files such as those downloaded from GenBank. Google tells me that Mac users can use TextEdit, but you need to be sure the format is plain text. I’m not a Mac person, so I don’t know for sure, but maybe someone can verify in the comments.
Since the annotation will list the amino acid sequence as part of the annotation, it is simple to copy and paste out the predicted protein sequence. The sequence of the annotated coding region, the CDS, which is the prediction for what might be a gene, is indicated by the base locations. The actual sequence of the CDS is part of the DNA sequence information as a whole. So, to get the specific sequence of CDS or a specific gene, we need to extract it out.
If we are working in a public database like NCBI GenBank, we can retrieve the sequence data or restrict it to just show the specific base range we need for the CDS. However, I am working with my own sequence information, which I have as the plain text, flat file of the annotation, in GenBank format. I could use a specialist program to open it and extract out the DNA sequence for the gene I need. But, I don’t have to do that. And, the purpose of this blog is to show you that you don’t need that and it can be done on any computer, with a bit of internet and some copying and pasting. No special software required.
In my case, I already know what sequence I want. I know from the annotation that it is present at “complement (226614..228458)”.
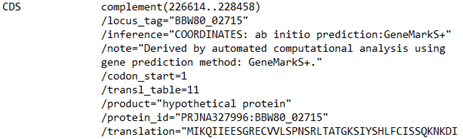
Interesting. The designation “complement” and the numbers in parentheses mean that the sequence of this predicted gene is going to be in the DNA in reverse complement. That means that the 226614 is going to be the end of the CDS and the 228458 is going to be the beginning. This is not what we might have expected, so something to watch out for when working with DNA sequences. Remember, the genes can be on either strand of the double helix. If the CDS was on the forward strand it would have just had the numbers to the right of the designation CDS without “complement” and the “()”.
I scroll through my file in Notepad and find the DNA and then the location of the CDS. It is possible then to highlight the CDS and copy the needed DNA sequence. Yes, this is the very low tech solution for doing this. There are other ways and programs that will achieve the same thing for you, but I wanted to show what can be done, by virtue of sequence files being plain text and therefore easy to manipulate on any computer.
Note that the first bases highlighted are TCA, which in reverse complement are TGA, a termination codon or stop codon. The last three bases are CAT, which would be the ATG reverse complemented initiation codon of the CDS. This is a good way to double check that you have the right area highlighted; check that you start and stop with an initiation and termination codon and that these are on the strand you expected based on the annotation.

If this was on the forward strand, I could paste it straight into a new Notepad file and make it a FASTA file of my own. But, it is in reverse complement, so I need to fix that first. There is a handy site to do this, which has not changed in many years: https://www.bioinformatics.org/sms/rev_comp.html. This does the basic operation needed.
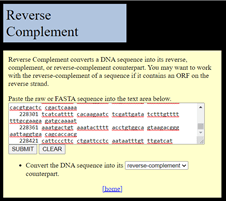
All of the non-DNA characters (the numbers from the annotated file) are deleted during the process. This is good. The output is the DNA in the right orientation, from the ATG start to the TGA stop.

The sequence is copied out of the Sequence Manipulation Suite output into a new Notepad file and made into a FASTA file through my addition of a line at the top that starts:
>Blog CDS for restriction digestion
Now that I am all set with the sequence of a gene – or in this case a sequence that has been predicted to be a gene – I can see which restriction enzymes might cut it.
The recommendation from Chapter 16 is to try NEBcutter:
I paste my sequence into the box to see which NEB enzymes will cut and get this output:

So, that’s the first part of Discussion topic 16.15 done. I have identified the enzymes that would cut the sequence. This on-line tool gives me a graphical output of the length of the CDS and shows where along it the various enzymes would cut. There’s more investigation that can be done from here, including zooming in and refining what is shown.
The Discussion topic wants me to identify those restriction enzymes that cut the CDS only once. This is easily done here on the NEBcutter tool. There is a link under the List heading that says “1 cutters”. I press that and all of the single cutters are listed, either alphabetically or by cut position.
The next parameters that the Discussion topic wants me to explore is to find:
- One enzyme that cuts at the start of the gene and generates sticky ends
- One enzyme that cuts at the end of the gene and generates sticky ends
- Both of the identified enzymes need to work at the same temperature
- Both of the identified enzymes need to work in the same buffer
To achieve the end goal I will change the sort order from alphabetical to cut position. Looking through the information on the first few, I am going to look first at XbaI. Why? This is an enzyme that I recognize and I think we have some in the freezer, so that will save on time and cost to use something we already have. I could pick anything and order something new, but I might as well use an enzyme from the freezer if it works for the experiment. Here the XbaI cuts T CTAGC with a 5’ overhang at 37°C in CutSmart Buffer at position 232/236.
Now I need one at the other end. There I find HindIII catches my eye, again as something we likely have in the freezer. It cuts A AGCTT with a 5’ overhang at 37°C in NEBuffer2.1 at position 1580/1584. That doesn’t look great, being in a different buffer from XbaI and at first I think I might need to look for a different enzyme on the list. However, there is more information on each enzyme on the product list page and I’ve just been looking at a summary.
Delving deeper, I see that HindIII has 100% activity in NEBuffer 2.1, but only 50% in CutSmart Buffer, so no help there. However, checking the product page for XbaI, I find out that it has 100% activity in CutSmart Buffer, but it also works at 100% in NEBuffer 2.1. So in NEBuffer 2.1 at 37°C I can do a double digest of the CDS under investigation with XbaI and HindIII, which will generate incompatible sticky ends and delete out 1348 bases of the coding region. I could then also digest an antibiotic resistance cassette marker with XbaI and HindIII to join with the cut ends and make selection of deletion mutants possible. Homologous recombination between the flanking regions brings the resistance marker into the chromosome, deleting the gene in the process, and the bacterial cells that are resistant to the antibiotic are those that have the gene deleted.
All of the digestion, ligation, and generation of the mutants would, of course, happen in a lab, but the planning of the experiments, such as investigating the presence of the restriction digest sites, can be done outside of the lab. Good planning of experiments is essential to making sure that experiments work well. Take the time now to plan experiments carefully. Think through protocols, write them out, and ensure they are really robustly planned, so that when you do go into the lab you have done everything you can to ensure success.

There are several other ways to achieve knock-outs and generate other mutations that are described in Bacterial Genetics and Genomics, as well as other uses for restriction enzymes. There are also great resources available on-line to support your use of the book, including slides with the figures from the book, like the one above, and flashcards to assist with learning terms.
That’s it for my blogs for Chapter 16, Gene Analysis Techniques. Next month I will get started on Chapter 17, Genome Analysis Techniques in my continuing theme on supporting research outside of the lab.
