Bacterial Genetics and Genomics, Chapter 10, Discussion topic 2.

In Chapter 10 of Bacterial Genetics and Genomics, the topic of genetics is explored and in particular of focusing on a single gene to determine its function. When we analyze a full genome sequence we assign names and potential functions to the regions that are assumed to be genes. But, these are just informed guesses. To really understand the bacteria, we must conduct experiments in the lab to investigate the function(s) of these genes. An annotation of a sequence in a genome could be completely wrong, it could be too specific and therefore misleading, or it could be correct. To find out we use one of our most powerful tools in genetic analysis, we generate a knock-out mutant so we can explore the loss of function of that gene.
In a recent article (https://journals.asm.org/doi/full/10.1128/mbio.02425-24), the function of a gene annotated as a phospholipase was investigated. It was observed that cells of Neisseria gonorrohoeae within neutrophil vacuoles tended to show dissolution of the vacuole membranes. It was hypothesized that there must be a lipase in the genome that was responsible for this phenotype. Three genes were identified as potentially being associated with this phenotype, phospholipase A (encoded by pla), phospholipase B (encoded by plb), and an alpha-hemolysin homologue (hylA).
To test whether there is a gene within N. gonorrhoeae that lyses membranes, as solid agar assay using red blood cells in the media was used. This showed that there is lysis activity. To test whether one of the three identified genes were involved, mutants were made in each of pla, plb, and hylA. There was a loss of function, a loss of the lysis activity, only in the pla mutant, identifying this gene as being responsible for the lysis activity.
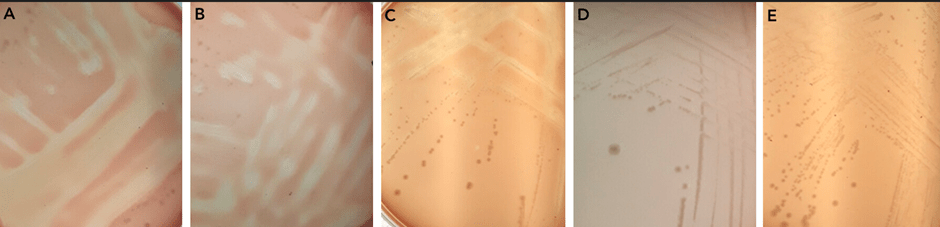
Areas of lysis of red blood cells in the agar plates are seen in panels A, B, and C. However, when pla is knocked out there is no red blood cell lysis as seen in panels D and E.
Using another powerful tool in genetic investigations, the pla gene was put into E. coli. There the E. coli gained the red blood cell lysis function, demonstrating that it is the pla gene responsible for lysis and not some other change in the N. gonorrhoeae pla mutant.
To further assess the function of the pla gene and confirm it was responsible for contact-dependent cell membrane lysis, two different mutants of pla were constructed. In addition, a different assay of lysis activity was used, using a fluorescent marker. As before, lysis was observed in the wild-type N. gonorrhoeae but not in the two different mutants of pla.
Although neutrophils are generally involved in eradicating bacteria from the human body, for N. gonorrhoeae these cells represent a safe haven. Within the neutrophil the gonococci can hide from the immune system and survive. To assess whether the phospholipase A is involved in this intracellular survival, neutrophils were infected with the wild-type and pla mutant N. gonorrhoeae. In both cases the bacterial count lowered initially, but for the wild-type the bacterial numbers recovered. There was no recovery for the mutant bacteria. Live/dead staining assays were able to show the number of live bacteria within the neutrophil compared to the dead bacterial cells.
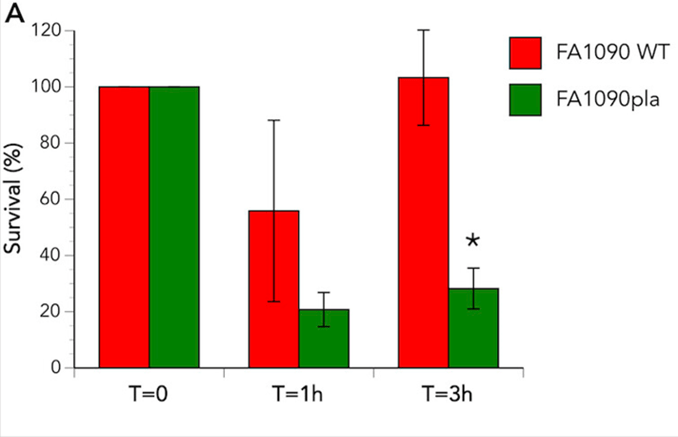
This bar graph shows the survival of bacteria within human neutrophils over time. In red are the results from the wild-type, which show 100% survival at time 0, between 50 and 60% survival at one hour, and recovery of the wild-type to 100% survival at three hours. In green are the results from the pla mutant, which show 100% at time 0, then a drop to about 20% at an hour, and significantly less recovery than the wild-type at three hours, where only about 25% of the mutants are viable in the neutrophils, compared to 100% in the wild-type.
Integrity of the neutrophil vacuoles was also assessed via electron microscopy, where the membranes of the vacuole can clearly be seen to the compromised in the wild-type. However, in the pla mutant the vacuole membrane appears intact. This provides even more supporting evidence of the activity of pla in lysing membranes and contributing to intracellular survival of the gonococcus.
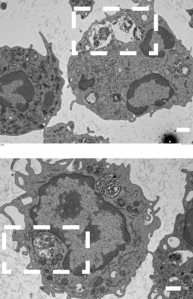
In the top panel, taken from an electron microscopy image, there is a clear region within the neutrophil cell where the N. gonorrhoeae bacteria are in a vacuole, which appears compromised. An uncompromised vacuole can be seen in the bottom image, where the gonococcal cells are contained within a neutrophil vacuole.
As you can see from this investigation, the generation of a knock-out mutant is a powerful tool in aiding our understanding of the function of a gene. With a mutant and is parent (the wild-type) to compare to one another, we can conduct a number of phenotypic assays to show that the loss of function has an impact upon the phenotype of the bacterial cell. In this case, phospholipase A loss results in a lack of ability to lyse host cell membranes and to therefore survive within neutrophils.
